| 所属 |
① 横浜市立大学 大学院生命医科学研究科 ② 横浜市立大学 大学院生命医科学研究科 |
|
|---|---|---|
| 氏名 |
① 池上 貴久 ② 元田 容子 |
|
| AMED 事業 |
課題名 | 生命科学と創薬研究に向けた相関構造解析プラットフォームによる支援と高度化 |
| 代表機関 | 理化学研究所 | |
| 代表者 | 山本 雅貴 | |
核磁気共鳴、重水素化蛋白質、メチル TROSY、相互作用解析
・安定同位体 2H, 15N, 13C で標識された高分子量蛋白質の調製
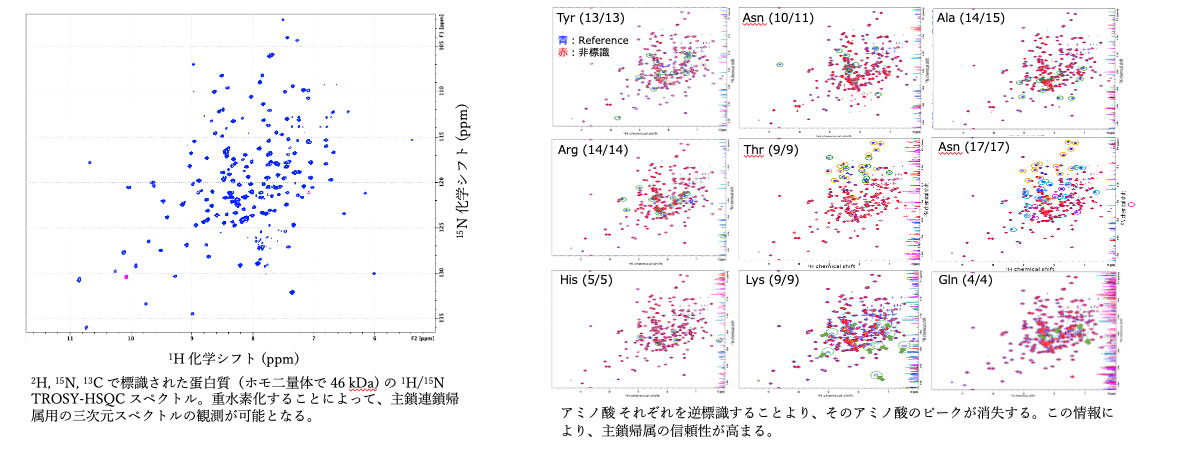
蛋白質の分子量が 30 kDa あたりを超えると、NMR ピークが非常にブロードとなり解析が難しくなる。しかし、側鎖の水素を重水素 2H 化すると、アミド基の 1HN, 13C, 15N のピークがシャープになり、70 kDa ぐらいまでは主鎖原子核の化学シフト値の帰属に従来の連鎖帰属法が使える。標識蛋白質の発現には大腸菌を重水の最少培地で培養するが、培地がかなり高価となるため、効率よく発現させるための M9 最少培地の作成法、培養法などが重要となる。また、ドメインごと、サブユニットごとに標識方法を変え、両蛋白質を後から intein や sortase を用いて融合させることもできる。このような試料を使うと、NMR の他、中性子線解析においても、観たい箇所のみを検出し、それ以外の背景を消すことができる。
・アミノ酸特異的非標識法
主鎖の化学シフト値の帰属のための一般的な手法である連鎖帰属法に加えて、アミノ酸特異的な標識ができれば、帰属の信頼性が非常に増す。そのためにしばしば 15N 標識アミノ酸が使われるが、アミノ酸種によっては高価である。そこで、13 種類のアミノ酸をそれぞれ特異的に非標識した安価な試料を調製する。大腸菌の代謝過程において他種アミノ酸への生合成(スクランブル)が起きるので、その情報も考慮に入れた帰属法を活用する。
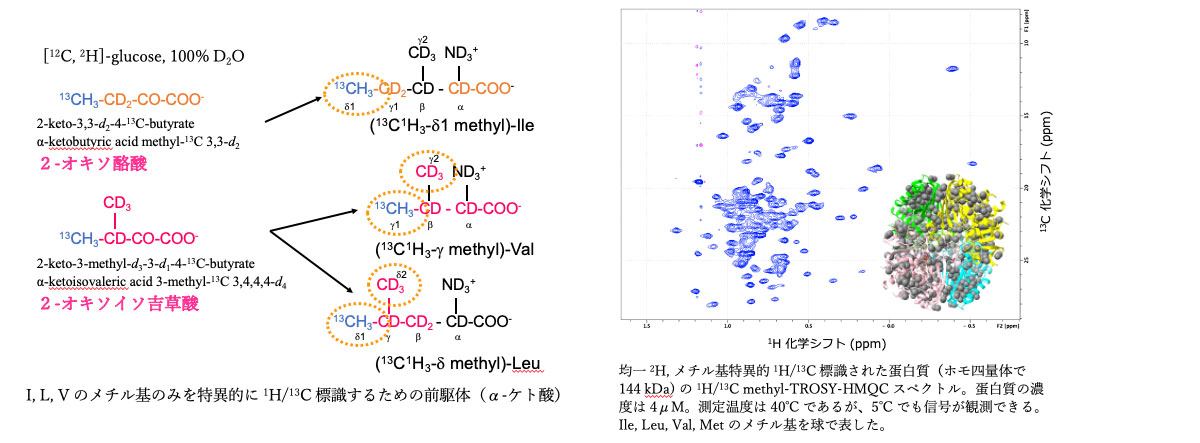
・メチル基特異的 1H/13C 標識、その他の部位を 2H で均一標識した蛋白質の調製
分子量が 50 kDa を超えると、アミド基 1H/15N をもとにした主鎖の帰属が困難となる。そこで、さらに感度の高いメチル基に着目する。具体的には Ile, Leu, Val, Met などのメチル基のみを特異的に 1H/13C で、それ以外を 2H/12C で標識した蛋白質を調製する。それには培地にそれぞれのアミノ酸前駆体(α-ケト酸など)を加える。そのための培地の作成、培養法などについて支援する。また、それらメチル基の帰属法としては、現在のところ、各残基を変異する方法がもっとも効率が高い。そこで、メチル基の数が多い場合は、優先順位を考慮して効率よく帰属を進めていく方法についても工夫が必要となる。
・複合体の立体構造決定
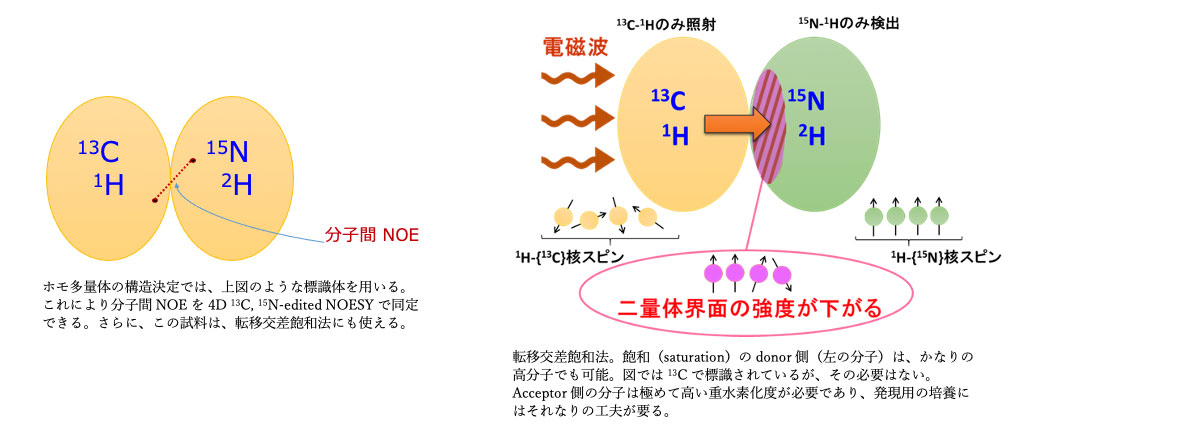
蛋白質単量体の NMR による構造決定法は比較的プロトコール化されているのに対して、複合体ではサンプル調製法も含めてかなり厄介である。さらにホモ多量体の場合は、同形のサブユニットでお互いに対応する核どうしの化学シフト値が重なるため構造決定はそれほど簡単ではない。そこで、安定同位体による標識法を工夫しながら、化学シフト摂動法、残余双極子相互作用(RDC)法、標識フィルター NOE 法、転移交差飽和(TCS)法などから得られる相互作用部位についての情報をドッキングソフト HADDOCK サーバに入力し、効率よくホモ多量体の構造モデルを構築する。
・相互作用解析
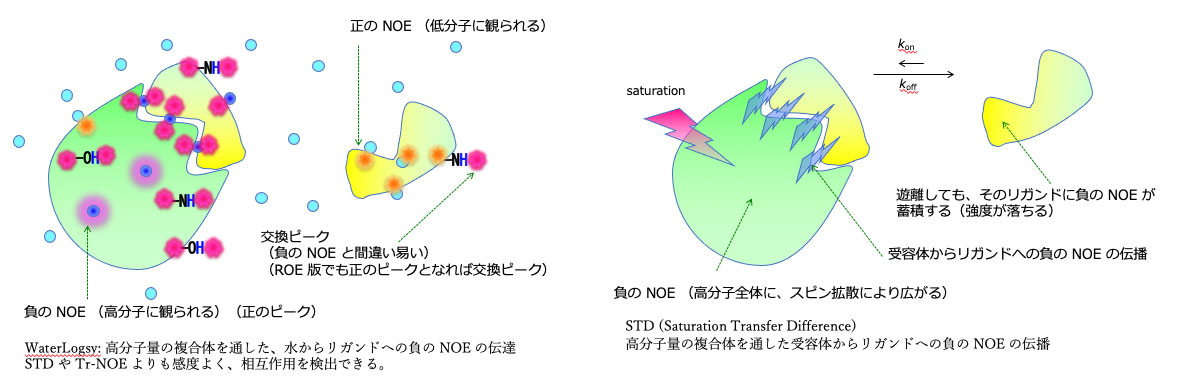
蛋白質どうし、蛋白質とリガンド(有機低分子、糖、セルロース・キチンなどの高分子、核酸)の相互作用を解析する。化学シフト摂動がもっとも簡単であるが、その他に上記の edited, filtered-NOE, transferred-NOE, 転移交差飽和(transfer-cross-saturation, TCS)法, Water-LOGSY法, saturation transfer difference (STD) 法などを駆使する。NMR は非常に幅広い親和性(Kd にして -6~-12 乗レベル)の相互作用を、その交換速度なども含めて解析できる。また、誤検知が少ない点も特徴的である。
・残余双極子相互作用(RDC)による方向情報の取得
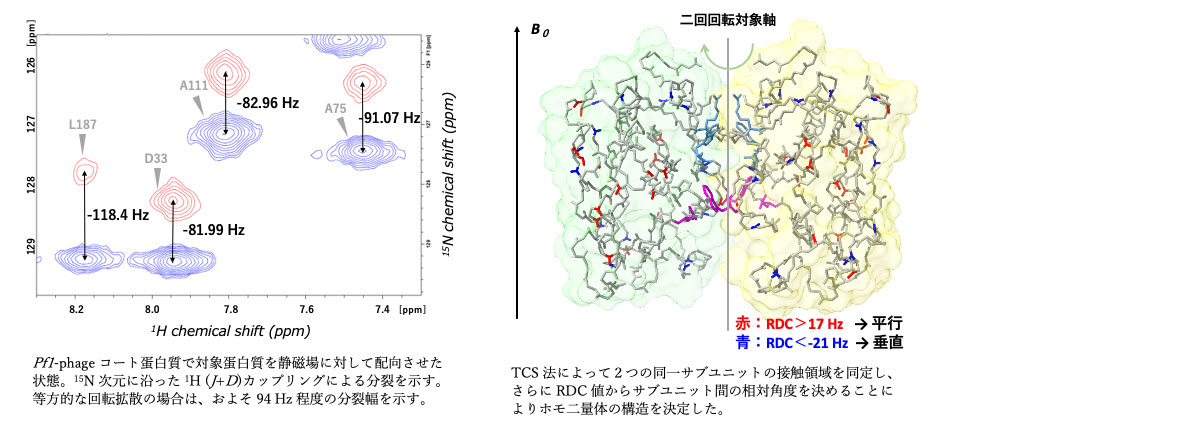
蛋白質を静磁場に対して配向させることにより、一般的な溶液 NMR では直接観測できない双極子間相互作用値をピーク分裂という形で復活させる。配向剤として、ポリアクリルアミドゲル、Pf1-phage コート蛋白質などを用いる。残余双極子相互作用(RDC)値をさまざまな方法で測定し、これを構造計算に用いる。特にホモ多量体の構造決定に活用すると、配向軸がホモ多量体の対称軸と重なるため、サブユニット間の相対角度を非常に精密に決めることができる。
・13C 直接測定
天然変性蛋白質(IDP)など非常にフレキシブルな領域を多く含む蛋白質の場合、1H 次元におけるピークの散らばりが非常に狭くなる。さらに高温、高 pH ともなれば、アミド基 1HN の溶媒との交換が速くなり、なおさら観測と解析が難しくなる。そのような場合は 13C 直接測定が有効である。当 NMR 施設においては、13C のプリアンプも冷却されているため、13C が感度よく直接観測できる。プロセスにおいてはホモ核 virtual decoupling など特別な処理が必要となる。
・蛋白質 A はホモ二量体(46 kDa)であるが、結晶の中ではパッキングのために複数の異なる向きをもった6量体を形成している。そこで、A を重水素化し、アミノ酸特異的非標識法も通して主鎖の NMR 信号を帰属した。その後、標識法の異なるサブユニットを混ぜ合わせ、サブユニット間の NOE, 転移交差飽和 TCS を通して、サブユニット界面を同定した。さらに、サブユニット間の相対角度を反映する RDC 値を測定し、それらの情報をもとに二量体の構造をドッキング計算した。
・蛋白質 B はホモ四量体(144 kDa)であり、40 種類ほどの他分子と相互作用することが知られている。しかし、複合体としての結晶例がない。これは、ひとつには相互作用が遷移的で、また動的に交換状態にあるためと予想される。そこで、NMR を使って他分子との相互作用を調べるために、B を重水素化し、さらに Ile, Leu, Val, Met のメチル基を 1H/13C で標識して methyl-TROSY-HMQC スペクトルを測定した。4 μM, 5℃ という NMR にとっては過酷な条件でも信号を観測することができた。さらに、Met 10 個と Ile 17 個に変異を入れることにより、それらのメチル基を帰属した。現在、さまざまな分子との相互作用を解析中である。
・転移交差飽和(TCS)法を利用して、キチン分解酵素と高分子量キチンとの相互作用、光合成で機能するフェレドキシンと巨大膜蛋白質複合体(光化学系 I、NDH 様複合体)との相互作用を解析した。このように TCS 法は超高分子量の相手分子との相互作用の検出にも適している。
・もともとは 2H, 13C, 15N などの安定同位体で標識した蛋白質の立体構造を NMR で決定することを専門とする。最近、蛋白質の構造は一般的に X 線結晶構造解析などで決定されることから、フレキシブルな分子の構造情報(動的構造)、弱い分子間相互作用、蛋白質と別分子リガンドとの相互作用などの解析にシフトしている。これらはなかなか結晶になりにくいことから、むしろ NMR が解析に向いていると思われる。
・相互作用解析では、最初に化学シフト値の変化を観ることが多いが、相互作用の相手分子が非常に大きい場合や相互作用が微かな場合は、むしろ転移交差飽和(TCS)法が解析に適している。今後は methyl-TROSY を利用した TCS 法に取り組むことにより、より大きな複合体を対象とする。交換状態における TCS をシミュレートするプログラムを開発したので、これを使って相互作用部位や交換速度などをより精密に見積もる。
・最近は、残余双極子相互作用(RDC)値があまり使われなくなった。しかし、RDC からは NOE などにはない遠距離に相当する構造情報が得られる。最近、50 kDa 以上の高分子量でも観測可能なパルス系列が開発された。そこで、これらを積極的に活用して、特にホモ多量体などの複合体の構造解析を進めていきたい。
・企業との共同開発では、Tr-NOE, Water-LOGSY, STD などを使って、蛋白質と創薬候補分子との相互作用を定常的に解析している。安定同位体で標識した蛋白質を使う場合とは異なり、これら非標識で少量の蛋白質を使いながらリガンド側を観測して相互作用の有無を判定するには、各種 NMR 測定パラメータの設定にかなりの経験が必要である。
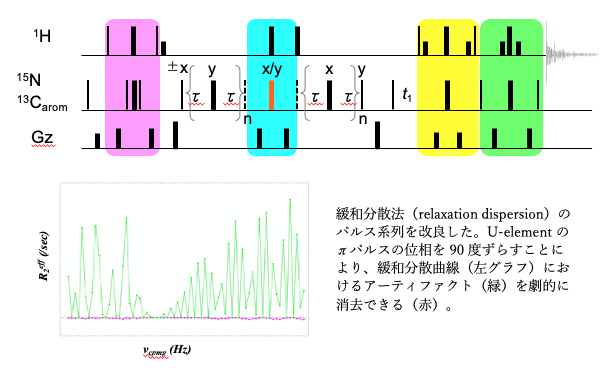
・緩和分散の測定をより少ないアーティファクトで精密に行うためのパルス系列を開発した。引き続き NMR の新規測定法の開発を続けていく。
・これまで、解析を進めるための各種ソフトウェアを数多く開発してきており、それらは支援でも活用していく。また、Mathematica や Python, Perl, C など便利な数値計算ライブラリが揃っているので、必要に応じて新たな機能をもったソフトウェアを即時開発していく。その一方で既存の良質の NMR 関連ソフトウェアの多くが使われずに埋もれており、それらを日常的に使っている経験者が希少になってしまった。現在、それらを自力で使いこなすことは初心者にとってはかなり難しいと思われる。そこで、できるだけ多くの研究者が、分野外からであっても、それらソフトウェアを研究に役立てられるように努めたい。